
Bromination of organic compounds is one of the most important transformations in organic synthesis. In addition to the many uses of brominated compounds as dyes, flame-retardants, and active pharmaceutical ingredients, organobromides serve as essential building blocks for the preparation of complex molecules.50 The good leaving group character of the bromide anion has made alkyl bromides a classical source of electrophilic synthons. Aryl bromides have also become highly important building blocks since the development of cross-coupling chemistry.
Introduction of bromine into organic molecules can be accomplished using elemental bromine (Br2). Br2 is an extremely corrosive and toxic reagent in both liquid and vapor form.51 Many other easy to handle brominating agents have been described,50N-bromosuccinimide being the most popular example. An early example of bromination using liquid Br2 in a microreactor was provided by the group of Hessel (Scheme 8a).52 The neat substrate was mixed with liquid Br2 using a micromixer, before entering a residence time unit which consisted of PTFE tubing. Solvent-free bromination of thiophene to produce the 2,5-dibromo derivative 11 (Scheme 8a) could be performed in less than one second with high selectivity and yields up to 86%. As the reaction is extremely fast (<1 s) important mixing effects were found on the reaction selectivity. Thus a selectivity of 88% was achieved using a micromixer with an internal active mixing geometry, while using a normal T-junction only 72% selectivity for the desired 2,5-dibromothiophene was obtained. Similar approaches have been used for the α-bromination of acetophenone53 and the 1,2-dibromination of alkenes.54Catalytic electrophilic bromination with Br2 and in situ generated FeBr3 was performed in continuous flow for the preparation of 2,4,5-trifluorobromobenzene 13 (Scheme 8b),55 an important intermediate for the synthesis of peptides and fluorescent reagents. The FeBr3 catalyst was generated by passing the Br2 solution through a PTFE tubing (i.d. 4 mm) packed with iron Dixon rings (0.69 × 0.69 mm, 0.2 mm diameter). The FeBr3 amount was controlled by the packed bed temperature and flow rate. At 70 °C and a flow rate of 0.2 ml min−1 a concentration corresponding to 10 mol% FeBr3was achieved. The solution was then mixed with the substrate using a T-junction before entering a reactor (FEP tubing) at 70 °C. After 4 min residence time the reaction mixture was quenched with aqueous Na2S2O3. A yield of 78% was obtained for the desired compound 13. Elemental bromine was used for the in situ generation of KOBr, a useful reagent for the bromination of methylsulfones by the group of Stevens (Scheme 8c).56 KOBr was rapidly generated upon mixing of neat Br2 with aqueous 5 M KOH. The resulting solution of KOBr in water was pumped and mixed with a solution of the substrate in toluene, generating a liquid/liquid segmented flow. Tetrabutylammonium bromide (TBAB) was used as ion exchange catalyst to enhance the reaction in the biphasic system.56 After 3 min residence time in a glass chip reactor at 85 °C quantitative yield and a productivity of >50 g per day for the desired tribromomethyl sulfones 14 was obtained.
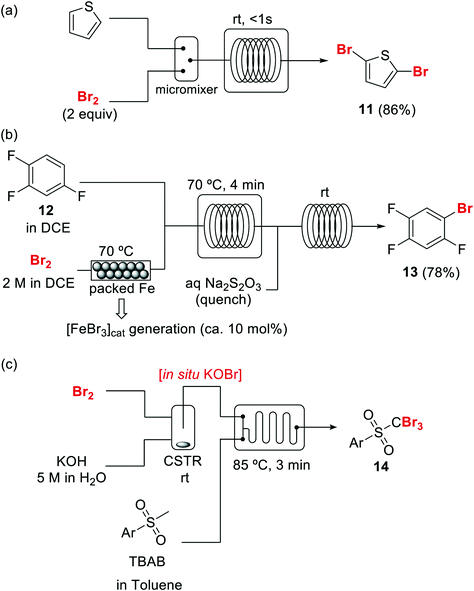
Scheme 8 Brominations with elemental bromine in flow (DCE = dichloroethane, CSTR = continuous stirred tank reactor).
Photochemical brominations using elemental bromine can be significantly accelerated in a microreactor, as demonstrated by the group of Ryu.57 Using a glass chip microreactor irradiated with a 360 nm LED (1.95 W) lamp a series of benzylic compounds could be brominated within seconds to minutes at room temperature (Scheme 9a). A single feed containing the substrate and Br2 (1.2 equiv.) dissolved in CCl4 was introduced into the photoreactor using a syringe pump. The reaction mixture was quenched at the reaction output by collection over an aqueous solution of Na2S2O3. Excellent selectivity for the monobrominated product was obtained. A similar approach using sun light to irradiate the flow reactor was reported by In and Park.58 Photochemical benzylic brominations with NBS in continuous flow have also been described.59,60 A green protocol using acetonitrile as solvent instead of chlorinated compounds such as CCl4 was achieved using a simple flow photochemical reactor built with transparent FEP tubing wrapped around a household CFL (Scheme 9b).59 With only 1.05 equiv. NBS a wide variety of benzylic compounds was converted to their corresponding bromides, with good to excellent yields being obtained in all cases. A throughput of 180 mmol h−1 was achieved. The same principle was used for the flow photochemical bromination to produce the 5-bromoethylpyrimidine 11, a precursor of Rosuvastatin, in a collaboration between scientist of Sandoz and the University of Ljubljana.60 Compared to batch mode, lower levels of side products and shortened reaction times were obtained. An important drawback of the use of NBS as brominating agent is the formation of equivalent amounts of succinimide as side product. Apart from complicating the isolation of the brominated products and formation of undesired waste, the presence of succinimide can create problems of clogging under continuous flow conditions, as it is insoluble in many organic solvents. An elegant approach to avoid these problems consists of extracting the succinimide into an aqueous phase using a liquid/liquid segmented flow regime, as demonstrated by O′Brien and Cooper.61 This method was applied to the bromination of enaminones 16 (Scheme 9c). An aqueous solution of Na2S2O3 and K2CO3 was mixed with the reaction mixture stream in dichloromethane. The excess of NBS was quenched and succiminide was extracted to the aqueous phase which could be conveniently separated using a liquid/liquid separator. The resulting organic phase contained the desired bromide 17 which could be isolated in good to excellent yields by simply evaporating the organic solvent. No traces of succinimide could be detected in the isolated products by 1H NMR.61
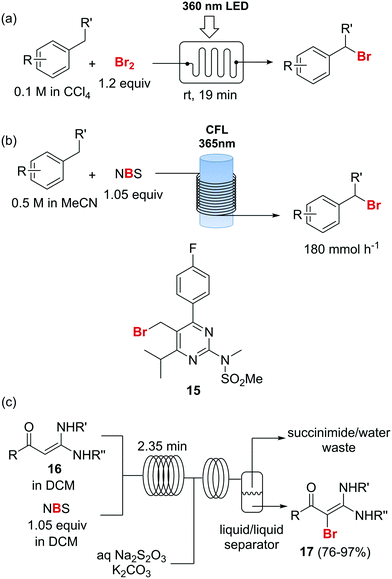
Scheme 9 Photochemical brominations and use of N-bromosuccinimide.
The preparation of vicinal 1,2-bromine azide derivatives can be achieved by treating olefins with bromine azide (BrN3). However, BrN3 is an extremely toxic and explosive compound, and its use in organic synthesis constitutes a very challenging task.62 A continuous flow protocol for the safe generation and use of BrN3 that enabled the preparation of vicinal 1,2-bromiazides was recently developed.63 The hazardous reagent was generated in situ using NaBr and NaN3 as bromine and azide sources, and Oxone as oxidizing agent. The continuous flow setup was based on a three-feed approach (Scheme 10). Two of the feeds contained aqueous solutions of NaBr and NaN3, and Oxone, respectively. A third feed contained the substrate 18 in an organic solvent (EtOAc or CH2Cl2). Upon mixing of the three feeds, a liquid/liquid segmented flow was created. The BrN3 rapidly formed in the aqueous phase was extracted into the organic phase containing the substrate. 1,2-Addition to the olefin was enhanced by irradiation with a black-light CFL (max. 365 nm) in continuous flow. The desired 1,2-bromine azides 19 were obtained with excellent purity, and could be isolated by simply separating the organic phase and evaporating the solvent. Good to excellent yields were obtained. A similar procedure was subsequently developed for the generation of chlorine azide (ClN3) and its addition to olefins.64
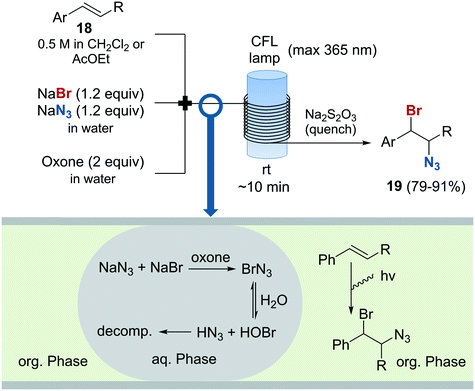
Scheme 10 Generation and use of bromine azide and its addition to olefins.


{kind=link}
{kind=link}
{kind=link}