Organofluoride compounds have become one of the most important and common class of substances among pharmaceuticals and agrochemicals (cf.Fig. 2b).22Decoration of an organic compound with fluorine atoms typically results in a significant enhancement of its biological properties.23 Increased bioavailability, lipophilicity, and metabolic stability otherwise difficult to obtain are typically acquired by the compounds after the introduction of one or more fluorine groups.23 The growing interest in this area has motivated intense research efforts for the development of novel catalytic methods and greener and more selective fluorinating agents.24 Elemental fluorine (F2) and hydrogen fluoride HF are among the most difficult to handle reagents in organic synthesis.25 F2 is the most powerful oxidant known, and both HF and F2 are highly corrosive and toxic compounds. Even some N-fluoro derivatives that are generally easier to handle are highly reactive, hazardous compounds. Fluorination reactions, especially those using F2 directly as fluorine source, are very exothermic and difficult to control. Lack of reaction selectivity is a common drawback. Not surprisingly, many research groups have studied the fluorination of organic materials using microreactor technology. The work in this area is extensive, and several reviews on the topic have been published.
Diethylaminosulfur trifluoride (DAST) is a widely utilized nucleophilic fluorinating agent. Its relatively high stability (it can be stored under inert atmosphere in the freezer for prolonged periods) and liquid form makes this reagent a convenient alternative for the lab-scale fluorination of alcohols, aldehydes and ketones. However, the use of DAST on large scale is very limited due to its propensity to detonate at temperatures above 90 °C. Under continuous flow conditions, the reaction exotherm can be readily dissipated and DAST fluorinations can be safely performed and scaled as demonstrated by the groups of Seeberger26 and Ley.27 In both cases the substrate and reagent in CH2Cl2 solution were mixed and reacted using fluoropolymer tubing as residence time unit, thus avoiding any degradation or corrosion of the reactor. At 70 °C several alcohols, carboxylic acids, and aldehydes were converted into the corresponding fluorinated derivatives after a residence time of 16 min (Scheme 1a).26 Unreacted DAST and the HF generated as side product were quenched inline using aqueous NaHCO3before the reactor output. Contact with HF was therefore totally avoided, increasing the safety of the procedure. An interesting application of this methodology was reported by Bayer Schering Pharma for the geminal difluorination of 17-keto steroid 1(Scheme 1b).28 The flow process could be performed safely with a productivity of 5–10 kg per day of the desired difluoro-derivative 2.
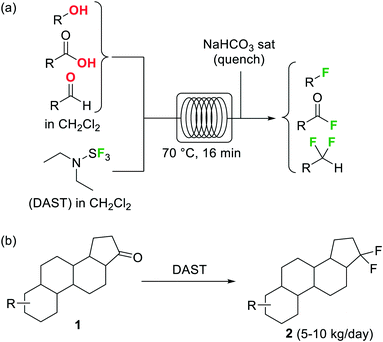
Scheme 1 Deoxyfluorination reactions using DAST in a microreactor.
Halogenation reactions, in which careful temperature control is typically required, perform well in continuous flow devices owing to their enhanced heat transfer. An illustrative example described by the Ley group is the diastereoselective electrophilic fluorination of the lithium enolate of carbonyl compound 3 with N-fluorobenzenesulfonimide (NFSI) (Scheme 2).29 The batch process required cooling of all reagents before addition at −78 °C. Simple addition of NFSI at room temperature to a −78 °C solution of the lithium enolate intermediate resulted in <50% conversion. Using a cryo-flow reactor at −60 °C, 88% conversion for the desired α-fluorocarbonyl product 4 (62% isolated) was achieved. Formation of the enolate from the substrate and LiHMDS could be performed at room temperature under continuous flow conditions.
Scheme 2 α-Fluorination of carbonyls using NFSI in a cryo-flow reactor.
Photochemical fluorination reactions, especially trifluoromethylations, have gained popularity during the past 5 years.25 Light induced reactions are significantly enhanced under continuous flow conditions due to the intense and uniform irradiation achieved within the capillary tubing in a flow reactor.30 In this context, various continuous flow trifluoromethylation protocols have been described.31 The direct fluorination of benzylic compounds 5 (Scheme 3) using Selectfluor as fluorine source and xanthone as catalyst using a photochemical flow reactor has recently been reported.32 The reactor consisted of a black-light (365 nm) compact fluorescent lamp (CFL) as light source and transparent fluorinated ethylene propylene (FEP) tubing. The combination of xanthone with black light irradiation resulted in a very fast fluorination process. Several benzyl fluorinated compounds were synthetized using this method, including biologically relevant examples 6a and 6b (Scheme 3).
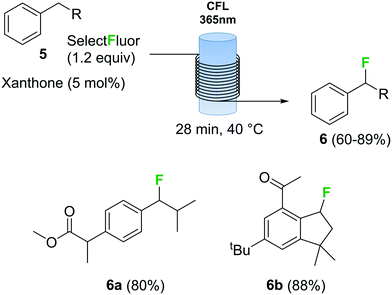
Scheme 3 Continuous flow photochemical fluorination of benzylic compounds with Selectfluor.
Fluorination of organic compounds using F2 as reagent is the preferred choice from the viewpoint of simplicity and atom economy. However, the extremely high reactivity of this reagent, in addition to the problems associated with its highly corrosive nature, are limiting factors for its implementation in organic synthesis. From the dawn of microreactor technology at the beginning of this century F2 fluorination of organic compounds has been approached by many research groups, including the groups of Hessel, Jensen, and the extensive contributions by Chambers/Sanford (see ref. 23 and references therein).33–38 In the pioneering work of Chambers and Spink, a microreactor fabricated from a block of nickel (or copper) specifically designed to perform fluorination reactions with elemental fluorine was described.36 The reactor showed excellent performance for the fluorination of β-dicarbonyl compounds (Scheme 4a). Scale-out of the microreactor using a simple numbering up strategy could be demonstrated for the same reaction using three reactors in parallel (Scheme 4b).37 In a more recent publication the microreactor was applied for the two-step synthesis of 4-fluoropyrazole derivatives 7 (Scheme 4c).38 After fluorination of the β-dicarbonyl compound, an organic hydrazine was introduced into the system through a T-mixer to generate the desired fluorinated pyrazoles in good yields.
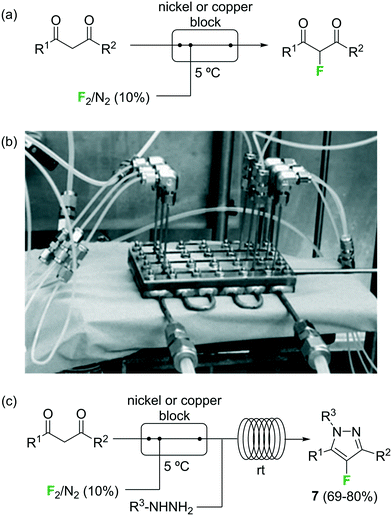
Scheme 4 Reactions with elemental fluorine. Image 4b reproduced with permission from ref. 37: R. D. Chambers, M. A. Fox, D. Holling, T. Nakano, T. Okazoe and G. Sandford, Lab Chip, 2005, 5, 191–198. Copyright© 2005, Royal Society of Chemistry.
Large scale fluorination of a 1,3-dicarbonyl compound commercialized by Axyntis was developed by scientist of the MEPI (Maison Européene des Procédés Innovants, France) using a sintered silicon carbide (SiC) reactor (Fig. 3).39 The excellent heat transfer capacity and corrosion resistance of the SiC material is perfectly suited for the highly exothermic reaction on a large scale. Using a ca. 2 M solution of the substrate in MeCN/AcOH (1 : 1) and F2/N2 in a gas/liquid flow regime excellent conversions were obtained resulting in a productivity for the desired product of approximately 200 g h−1.
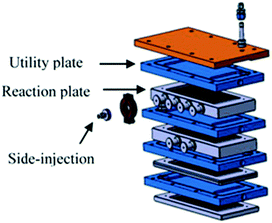
Fig. 3 Continuous flow SiC reactor. Reproduced with permission from ref. 39: S. Elgue, A. Conte, C. Gourdon and Y. Bastard, Chim. Oggi, 2012, 30(4), 18–21. Copyright© 2012 Teknoscienze Publisher.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}