Knowledge of the geochemical distribution of elements involves elucidation of the relative and absolute abundances of the chemical elements in the Earth and in its various parts—the crust, interior, atmosphere, and hydrosphere. This comprises a major part of the science of geochemistry, which is the study of the distribution of the chemical elements in space and time and the laws governing this distribution. Basic knowledge in this area was largely accumulated during the 19th century. As noted above, the concept of a limited number of chemical elements had been established by 1800, and the appearance of the periodic table, in 1869, provided a new insight into the limitations on the number of elements. Concurrent with these advances in chemical understanding, from about 1850 onward there was a steadily increasing output of analytical data on the Earth’s rocks, minerals, and waters, mainly from laboratories in Europe and North America. The output from North America was materially increased following the establishment of the United States Geological Survey in 1879 and the appointment of Frank W. Clarke as chief chemist in 1884.
Clarke’s name will always be linked with the study of the geochemical distribution of the elements—indeed, the term clarke was proposed as the unit for the average percentage of an element in the Earth’s crust by Soviet scientists and has been generally adopted. In 1889 Clarke wrote the first of his many publications on the geochemical distribution of the elements. He assembled many chemical analyses of rocks from different continents, calculated average values, and showed that the overall chemical compositions of continental areas are remarkably similar. By combining these averages he obtained values for the abundances of the commoner elements in the continental crust of the Earth, values that have not been materially changed in spite of the vast increase of available data since that time. He also estimated abundances for many of the less common elements; these estimates were based in many instances on very limited and imprecise data and subsequently have been improved.
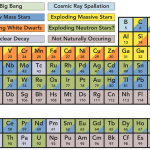
A further development of great significance was the assemblage of comprehensive data on the abundances of individual elements in terrestrial materials and in the Cosmos (based on solar and meteorite abundances) by the Norwegian geochemist Victor Moritz Goldschmidt during the 1930s. Goldschmidt’s tables provided the basis for modern research on the geochemical distribution of the elements, and his compilation of data on cosmic abundances was the key to later theories on element synthesis in stars and supernovae.
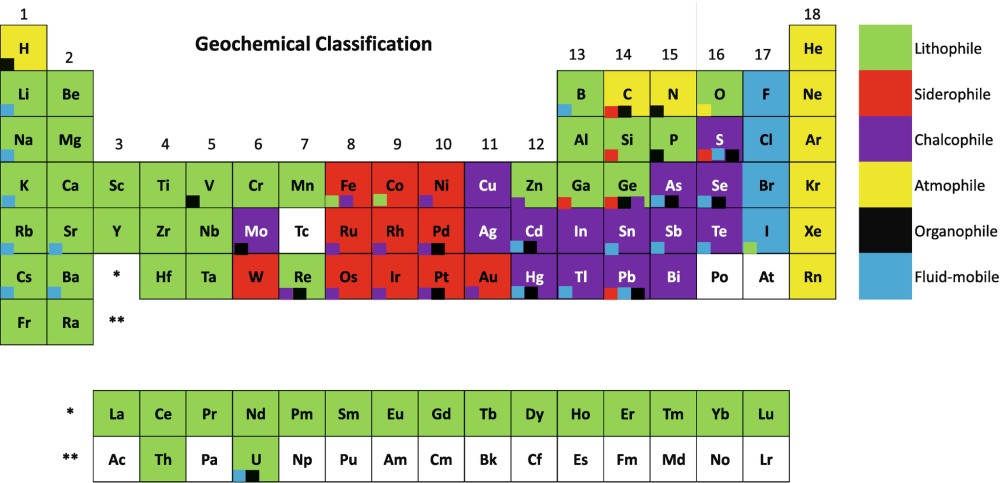
Goldschmidt also contributed to the understanding of elemental distribution within the Earth through his geochemical classification of the elements into lithophile, siderophile, chalcophile, and atmophile. Lithophile elements are those with a strong affinity for oxygen; they are concentrated in the crust or lithosphere as silicate and oxide minerals. Siderophile elements are principally metals that alloy readily with iron; Goldschmidt explained their scarcity in the Earth’s crust by their concentration in the nickel–iron core (the siderosphere). Chalcophile elements are those with strong affinity for sulfur; they occur mainly as sulfides. And atmophile elements are gases, such as nitrogen, argon, and other rare gases, which are unreactive and hence accumulate in the atmosphere. (Goldschmidt also proposed a group of biophile elements, for those that concentrate in living matter—essentially carbon, hydrogen, oxygen, nitrogen, sulfur, and phosphorus.)
Terrestrial distribution
The study of earthquake waves passing through the body of the Earth has shown that the interior is not uniform; it consists of distinct shells separated by concentric discontinuities at which the velocities of the passing waves change. The two major discontinuities that are universally recognized are the Mohorovičić Discontinuity, which divides the Earth’s crust from its underlying mantle, and the Wiechert–Gutenberg Discontinuity, which separates the mantle from the core. The latter discontinuity exists at a depth of 2,900 kilometres (1,800 miles); it is marked by a sudden increase in density, from about 5.7 at the base of the mantle to 9.7 at the top of the core. The only reasonable interpretation of this discontinuity is that the mantle consists of silicates and oxides of the common elements (largely magnesium and iron), and the core consists of metallic iron alloyed with minor amounts of other elements (analogous to the nickel-iron in meteorites). The Mohorovičić Discontinuity varies in depth from place to place; it averages about 33 kilometres (20 miles) below the continents and about 8 kilometres (5 miles) below the bottom of the deep oceans. It too is marked by a density increase from crust to mantle—a comparatively small one, from about 3 to 3.3.
To the three spherical divisions—crust, mantle, and core—two more should be added: the hydrosphere, which is the discontinuous shell of fresh and salt water, on and within the crust; and the atmosphere, the ocean of air that surrounds the Earth, gradually thinning into the vacuum of outer space.
The Earth’s core
The evidence for the composition of the core is all indirect because no means have yet been devised for directly sampling the deep interior of the Earth. The moment of inertia of the Earth indicates that there is a concentration of mass around the centre, and seismic data have shown that below the Wiechert–Gutenberg Discontinuity the density of the material is high, ranging upwards from 9.7. The only heavy element with high cosmic abundance is iron, and because an iron–nickel alloy is an important meteorite component, it is reasonable to conclude that the Earth’s core consists largely of metallic iron with a minor admixture of other elements. This conclusion is supported by geophysical evidence that indicates that the mean atomic number of the material of the core is about 22. The atomic number of iron is 26, so this implies that the core also contains elements of lower atomic number. Sulfur, with atomic number 16, and carbon, 6, are relatively abundant in meteoritic matter, and the presence of minor amounts of these elements in the core would effectively reduce the mean atomic number. Some authorities have advocated silicon (atomic number 14) as the major alloying component in the core, but this seems less likely; if silicon were the sole alloying element, then the core would have to contain more than 30 percent silicon in order to reduce its mean atomic number to 22. In addition, free silicon requires extremely reducing conditions (lack of oxygen), and the presence of ferrous iron in the mantle is inconsistent with this requirement.
It is not possible to give definite figures for the abundances of the elements in the Earth’s core. It is certainly made up largely of metallic iron, however, probably with some nickel, a little cobalt, and appreciable amounts of such lighter elements as carbon and sulfur.
The Earth’s mantle
The mantle comprises that part of the Earth between the Mohorovičić and the Wiechert–Gutenberg discontinuities. It makes up 83 percent of the volume of the Earth and 67 percent of its mass and is thus of decisive importance in determining the bulk composition of the planet. In estimating elemental abundances in the mantle, however, the same difficulty as with the core arises: direct sampling is not feasible. Much more geophysical data are available for the mantle, however, and some volcanic eruptions have brought rock fragments to the surface that have certainly been derived from this zone. The most remarkable of these materials are the diamond-bearing inclusions found in the famous pipes, or volcanic necks, that are mined in South Africa and Siberia. The presence of diamond, the high-pressure form of carbon, implies a depth of origin of at least 100 kilometres (62 miles), but these inclusions are rare. The common type of mantle-derived inclusion is peridotite, a silicate rock consisting largely of olivine, (Mg,Fe)2SiO4, with minor amounts of orthopyroxene, (Mg,Fe)SiO3, and diopside, CaMg(Si2O6).
Geophysical information indicates that below a depth of about 1,000 kilometres (620 miles), the mantle behaves as an essentially homogeneous material, but above this level its physical properties are more varied, and there is evidence for second-order discontinuities. This region above 1,000 kilometres is frequently referred to as the upper mantle, and in recent years has been the object of a concentrated research effort by geologists and geophysicists all over the world. The significance of the upper mantle is that processes originating there have dramatic effects on the surface—in the form of volcanic eruptions and some earthquakes—and less dramatic but equally important effects within the crust, such as the introduction and concentration of some elements, possibly leading to the formation of ore deposits. Increased knowledge of the upper mantle thus has both scientific and economic appeal.
Geophysical data on the properties of the upper mantle suggest that it must consist essentially of magnesium-iron silicates, probably largely olivine in the region immediately below the crust. Olivine is not stable under very high pressures, however; it is converted to a different phase of about 10 percent higher density and with a structure like the mineral oxide spinel (MgA12O4). This conversion would occur in the mantle at depths of around 400 kilometres, and a second-order discontinuity at that depth can plausibly be ascribed to this conversion. Pyroxenes also undergo transformations to phases of greater density at the high pressures within the mantle. Thus the mantle, although composed of material of familiar chemical composition, consists, in its lower part at least, of different minerals than those in the upper part.
Many estimates of the composition of the upper mantle have been made in recent years. On the whole, the similarities are more important than the differences. All agree that the principal components are oxides of silicon, magnesium, and iron. The differences are mainly in the minor components such as aluminum oxide, calcium oxide, and the alkalies, and are determined largely by theoretical considerations and the weight given to specific aspects of the geophysical and geochemical data.
Although fairly reliable estimates exist for the abundances of the major elements in the mantle, little is known of minor and trace elements. Knowledge of the crystal structure of possible mantle minerals indicates that many minor and trace elements will not be readily incorporated, however. They are therefore likely to concentrate in liquid material in the mantle and be carried upward in solution, eventually being transported into the crust. It is thus probable that the mantle is relatively depleted, and the crust relatively enriched, in minor and trace elements. This is certainly true for uranium and thorium, because the amount of these elements in the crust is almost sufficient to account for the total amount of heat flowing out of the Earth.
The Earth’s crust
The crust is a comparatively thin shell on the surface of the Earth and makes up less than 1 percent of its total mass. Its geochemical significance is only marginally related to its bulk, however. It has been subjected to extensive investigations, and it provides the raw materials on which civilization depends. It is the most diverse of the geospheres, being a complex mosaic of many rock types—igneous, sedimentary, and metamorphic—each with a wide variety of chemical and mineralogical compositions. The surface is veneered with soils, related in composition to the rocks from which they formed, but with important modifications because of the smaller grain size, the presence of organic matter, and an intricate complex of living organisms. Ultimately, man’s welfare and indeed his survival depends on the wise utilization of the materials in the crust. Modern civilization has been erected upon the exploitation of fuels and ore deposits, which are simply geochemical concentrations of useful elements.
Igneous rocks
Clarke estimated that 95 percent of crustal rocks are of igneous origin (formed from molten silicate masses, or magmas). Sedimentary rocks occur as a thin veneer on an igneous or metamorphic basement, except where locally thickened in mountain belts. The primordial rocks of the crust must have been essentially igneous, and the first sedimentary rocks were derived from them by processes of weathering and erosion. Metamorphic rocks are formed from both sedimentary and igneous rocks by transformations due to heat and pressure at depth in the crust; unless very intense, these transformations do not totally obliterate the primary igneous or sedimentary features.
Major components
Igneous rocks show a wide range of composition; the principal component, silica (SiO2), ranges from about 35 percent to 80 percent among the commoner igneous rocks, and other components also show a wide variation. They thus illustrate some quite extensive geochemical fractionations of the elements, the fractionations that may have economic significance if they bring about the findings of workable ore deposits.
In 1924 a comprehensive review of igneous rock composition based on compilation of over 5,000 superior analyses was published. This was in many respects the ultimate refinement of Clarke’s initial review of 1889. It confirmed that the averages of analyses from different continental areas are essentially identical. It also revealed significant geochemical differences between the continental and oceanic crusts. The average of igneous rock analyses from the oceanic islands is notably lower in silica and alkalies, and higher in magnesium and calcium oxides, than the continental averages. This is simply a reflection of the fact that most oceanic islands, such as Hawaii, consist almost entirely of basalts (averaging about 50 percent silica), whereas continental areas include large granitic masses, with silica contents around 70 percent. In terms of volumes, igneous rocks consist predominantly of two great types, granitic and basaltic. The former essentially are confined to the continents and the latter occur in both continents and ocean basins. The other types of igneous rocks, while many and varied, are quantitatively insignificant and hardly affect the averages. Thus, for the major elements, the average of over 5,000 analyses of igneous rocks is not significantly different from the simple average of two individual rocks, a granite (G-1) and a basalt (W-1). This can be seen from the Table, by comparing G-1 and W-1 values with those in the column headed “Earth’s crust.”
The figures for the specific granitic (G-1) and basaltic (W-1) rocks are included in the Table because they have been analyzed for practically all the elements in different geochemical laboratories throughout the world. The rock G-1 was a granite from Westerly, Rhode Island, and W-1 was a basaltic rock (specifically a diabase) from Centerville, Virginia. Several hundred kilograms of each of these rocks were crushed to a fine powder in the laboratories of the U.S. Geological Survey and samples distributed to analytical laboratories throughout the world, in order to obtain as many analyses as possible. The results were then critically examined, and the figures given in the Table are considered to be the best available in terms of accuracy and precision. G-1 and W-1 are undoubtedly the most thoroughly analyzed rocks and now serve as basic geochemical standards. It must be borne in mind that they are individual rocks, however, and cannot be considered to be averages of all granites and all basalts. Indeed, it is clear that G-1 is unusually rich in some trace elements (e.g., thorium).
Perhaps the most significant feature of the composition of the Earth’s crust is that it is dominated by comparatively few elements. Only eight—oxygen, silicon, aluminum, iron, calcium, magnesium, sodium, and potassium—are present in amounts greater than 1 percent, and these eight make up almost 99 percent of the whole. Of these, oxygen comprises almost 50 percent by weight. The dominance of oxygen is even more marked when weight percentages are converted to atomic percentages, as follows: oxygen (weight percentage, 46.6; atomic percentage, 62.2), silicon (27.7; 21.2), aluminum (8.13; 6.47), iron (5.00; 1.92), calcium (3.63; 1.94), magnesium (2.09; 1.84), sodium (2.83; 2.64), potassium (2.59; 1.42). This comparison, of course, merely emphasizes the fact that the crust consists almost entirely of oxygen compounds, mostly silicates and aluminosilicates of iron, calcium, magnesium, and the alkali metals. As Goldschmidt remarked, the lithosphere may well be called the oxysphere. Clarke and his collaborators calculated that the average mineralogical composition of igneous rocks is: quartz 12.0 percent, feldspars 59.5 percent, pyroxene and hornblende 16.8 percent; biotite 3.8 percent, titanium minerals 1.5 percent, apatite 0.6 percent, and other accessory minerals 5.8 percent.
Elements of minor and trace abundance
So far only the major elements have been discussed, those present in amounts greater than 1 percent by weight. Consideration must now be given to the minor and trace elements. The data are given in the Table and are presented diagrammatically in theFigure.
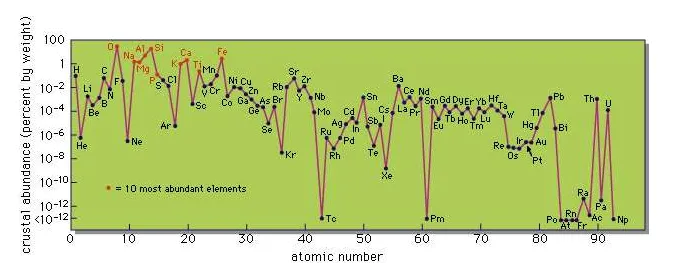
Crustal abundances of elements of atomic numbers 1 to 93.
A cursory examination immediately reveals some intriguing features in the abundance pattern. The predominance of the even-numbered elements over the neighbouring odd-numbered ones is still apparent but not so regular as in the cosmic abundances. Chemical fractionations taking place during the evolution of igneous rocks from primordial matter have clearly modified this basic relationship. The odd–even relationship is most prominent in the rare-earth elements (atomic numbers 57–71), which are chemically so similar that they are little fractionated by geochemical processes.
A particularly noteworthy feature is that some unfamiliar elements, such as rubidium, are relatively abundant, whereas others, such as most of the industrial metals except iron and aluminum, are actually of very low abundance. Thus boron, familiar to every homemaker in the form of borax cleansers and boric-acid antiseptics and well-known in the ancient world, is an element of extremely low abundance, much lower, for example, than zirconium, and lower even than hafnium, an element first discovered as late as 1923. Mercury, another element known to the ancient Greeks and Romans, is also of extremely low abundance. On the other hand, the so-called rare-earth elements are not extremely rare. Evidently one must make a clear distinction between abundance and availability. Some elements may be of low abundance but are readily available, because geochemical processes result in their concentration in specific deposits which can be commercially exploited. Other elements that are relatively abundant may be widely dispersed in small amounts and seldom or never occur in concentrated large deposits. A typical example is titanium, which is present in practically all igneous rocks in amounts ranging up to several percent, but which, in spite of its useful properties as a metal, is still not widely used. This is partly because of its dispersed nature and partly because of the technical difficulties in extracting the element from the minerals in which it occurs.
The distribution of minor and trace elements in igneous rocks, for those of lithophile affinity, is largely controlled by their ionic radii or size. Minor and trace elements with radii similar to those of major elements can substitute for these elements in the common minerals of the igneous rocks. The crystal structures of these minerals act as sorting mechanisms, accepting those atoms of appropriate size and rejecting others. Thus rubidium, with a radius of 1.47Å (one angstrom [Å] = 10−8 centimetres) is incorporated in potassium feldspar, KAlSi3O8, because its radius is close to that of potassium (1.33Å). The next higher alkali element, cesium, with a considerably larger radius (1.67Å), is not accepted into the feldspar structure; it remains in the igneous liquid during the crystallization of the major minerals until its concentration increases to such an extent that it can form the independent mineral pollucite (CsAlSi2O6).
The factor of ionic size, coupled with geochemical affinity (lithophile, chalcophile, or siderophile) is a key to the distinction between abundance and availability. Elements that are similar in size and geochemical affinity to major elements are dispersed in small amounts in common minerals; i.e., rubidium in potassium feldspar, gallium in aluminum minerals, and germanium in silicate minerals. Elements that do not readily enter the common minerals of igneous rocks remain in the residual melt as crystallization proceeds. Fractional crystallization of magmas (igneous melts) normally results in a residual liquid of granitic composition. Under suitable conditions this residual liquid solidifies as a coarse-grained rock known as a pegmatite. Pegmatites are famous for their content of rare and unusual minerals, which contain many of the minor and trace elements. They are the commercial sources of lithium, beryllium, scandium, yttrium, the rare earths, cesium, niobium, and tantalum, all elements that concentrate in the residual liquid because of their specific geochemical properties.
Chalcophile elements are all of rather low abundance, and the minerals that they form, mainly sulfides and some arsenides, are not stable at the high temperatures of igneous crystallization. Sometimes these elements are found in granites and pegmatites—molybdenite (MoS2) is a typical example. More frequently they are removed from the crystallizing magma as hot aqueous solutions and are deposited as metalliferous veins in the surrounding rock. Sometimes they may reach the surface as components in thermal springs; mercury has been deposited (as native mercury and as cinnabar, HgS) by some of these springs, occasionally in sufficient amounts for profitable mining.
Sedimentary rocks
The decomposition of pre-existing rocks by weathering, the transportation and deposition of the weathering products as sediments, and the eventual formation of sedimentary rocks might be expected to produce a gross mixture of materials, thereby working against further geochemical differentiation of the elements. This is not the case; sedimentary processes frequently produce remarkable concentrations of the elements, leading to almost pure deposits of certain minerals. Some sandstones, e.g., contain over 99 percent quartz and some limestones over 99 percent calcium carbonate. The ultimate is reached in salt deposits, with extensive beds of anhydrite (CaSO4), gypsum (CaSO4 · 2H2O), halite (NaCl), and other compounds. Goldschmidt compared the sedimentary process with a quantitative chemical analysis, involving the successive separation of specific elements or groups of elements.
Quartz (SiO2) is highly resistant to weathering and accumulates as deposits of sand. When consolidated these deposits form sandstones, an important group of sedimentary rocks. Under special conditions almost any mineral may be deposited in sand-sized grains, but most minerals are eventually decomposed by weathering. A few resistant ones may survive and be sufficiently concentrated to form economic deposits known as placers; the most familiar are probably the gold-bearing sands, important sources of this element, but sand deposits may have economic concentrations of zirconium (as the mineral zircon, ZrSiO4), titanium (as rutile, TiO2, and ilmenite, FeTiO3), tin (as cassiterite, SnO2), and others.
The aluminosilicates of igneous rocks, mainly the feldspars, (K,Na)AlSi3O8 and (Na,Ca)(Al,Si)4O8, are relatively easily decomposed by weathering. The alkali elements and calcium are largely carried away in solution, whereas the aluminum and silicon are quickly redeposited as insoluble clay minerals. When consolidated, these minerals form shales and mudstones. The ferromagnesian minerals undergo a more complex decomposition, sometimes leading to the deposition of iron-rich sediments consisting largely of hydrated ferric oxide; such sediments are valuable iron ores in many countries.
Calcium is carried away in solution mainly as calcium bicarbonate, Ca(HCO3)2. Most of it eventually reaches the sea, where it is utilized by a vast variety of organisms as skeletal material in the form of calcite and aragonite (polymorphs—different forms—of CaCO3). Accumulation of skeletal materials after death of the organisms has formed extensive deposits of limestone throughout geological time. Magnesium in seawater can react with calcium carbonate to form dolomite, CaMg(CO3)2, and in this way some magnesium is removed from solution and deposited in sediments.
Much of the magnesium, however, remains in seawater, which is essentially a dilute solution of magnesium, calcium, sodium, and potassium chlorides and sulfates, with many other elements in small amounts (see Table). Under special geological circumstances bodies of seawater can be cut off from the open ocean, and under arid conditions the water will evaporate and extensive salt beds be deposited. Such conditions have occurred in different regions throughout geological time, and the resulting salt deposits are economically important as sources of sodium, potassium, calcium, magnesium, chlorine, and sulfur.
The three major groups of sedimentary rocks are sandstones, shales, and the carbonate rocks (limestones and dolomites). Much less geochemical research has been devoted to sedimentary rocks than to igneous rocks, and the data for their contents of minor and trace elements are therefore less extensive. The figures in the Table show that the minor and trace elements generally tend to be more concentrated in shales than in the sandstones and carbonate rocks.
The problem of arriving at an average composition for all sedimentary rocks is still largely unresolved, largely because of uncertainty in the relative amounts of shales, sandstones, and carbonate rocks. From geochemical arguments Clarke estimated the relative percentages of these three groups as 80:15:5, respectively. Actual measurements of sedimentary rocks suggest that these figures overestimate the amount of shales and underestimate that of limestones, however. Thus, a compilation of the recorded amounts of shales, sandstones, and limestones in more than 213,000 metres (700,000 feet) of sedimentary rock formations gave relative percentages of 46:32:22, respectively. The identification of a formation as a limestone, a sandstone, or a shale, however, is likely to be gross; shales usually contain considerable sand, sandstones may carry much clay, and the term limestone is applied to many rocks with 50 percent or less of carbonate. It does appear that limestones are more prominent in the geological record than might be expected from geochemical calculations, however; this probably reflects the fact that shallow-water environments are the great places of carbonate deposition, whereas the ocean deeps are the repository primarily of clay-rich sediments.
Metamorphic rocks
Comparatively few investigations have been made of the elemental composition of metamorphic rocks. Many of these rocks retain the geochemical features of their parent igneous or sedimentary materials, and their bulk composition has been little changed despite complete recrystallization and the production of new minerals and structures in some instances. Some metamorphic rocks, however, have been markedly modified by the removal of some components and the addition of others.
The Geological Survey of Canada has performed a comprehensive study of a large area of the Canadian Shield, a region of complex geology largely made up of metamorphic rocks. From a collection of more than 8,000 bedrock samples, the average abundances of all the major elements and a number of minor and trace elements were determined; the figures are given in the Table. As might be anticipated, the average composition is not very different from the average composition of igneous rocks. It does show a somewhat higher silicon content, probably reflecting a preponderance of granitic over basaltic rocks and a relative abundance of quartz-rich sedimentary rocks in the original makeup of the Canadian Shield. The general validity of these abundance figures for metamorphic rocks has been confirmed by a similar study of the average composition of metamorphic rocks in the former Soviet Union, which has given closely comparable results.
Ore deposits
An ore deposit, in its simplest terms, is a portion of the Earth’s crust from which some industrial raw material can be extracted at a profit. As such, its characteristics are as much economic as geochemical. Nevertheless, its formation required the operation of geochemical processes to produce the concentration of a specific element or elements in a particular place. Economics decide whether this concentration is rated as an ore deposit or merely as a deposit of scientific interest. The economics may change with time, depending upon price, availability of transportation, cost of labour, and other factors.
Some general principles can, however, be enunciated. Proceeding from the average abundance of an element in the crust, and the minimum abundance that can be profitably exploited under normal circumstances, a factor of enrichment necessary to produce an ore deposit can be derived (see Table). The economic control is immediately evident in the approximate relation between the factor of enrichment and the price of the product sought. The most extreme example of this is in diamond mining, where the product sought may be present in the rock mined in as low a concentration as 1 part in 50,000,000. Ease of extraction, of course, plays an important role in this. Diamonds are readily separated from the great mass of waste rock by a relatively simple and inexpensive process. Magnesium is commercially extracted from seawater, where its concentration is 0.13 percent, rather than from the common rock dunite, where its concentration is about 25 percent, because of the ready availability of seawater and the cheapness of the extraction process.
| Concentration factors for ore bodies of common metals | |||
| metal | percent in Earth’s crust | minimum percent profitably extracted | enrichment factor necessary for an ore body |
| aluminum | 8.13 | 30 | 4 |
| iron | 5.00 | 30 | 6 |
| manganese | 0.10 | 35 | 350 |
| chromium | 0.02 | 30 | 1,500 |
| copper | 0.007 | 1 | 140 |
| nickel | 0.008 | 1.5 | 175 |
| zinc | 0.013 | 4 | 300 |
| tin | 0.004 | 1 | 250 |
| lead | 0.0016 | 4 | 2,500 |
| uranium | 0.0002 | 0.1 | 500 |
Ore deposits may be found in all types of rocks—igneous, sedimentary, and metamorphic—and seawater is also a significant source of such elements as sodium, chlorine, magnesium, and bromine. There are many processes of geochemical enrichment leading to the formation of ore deposits, and they are often the end result of a complex series of such processes acting over a long period of time. The economic importance of ore deposits has ensured their thorough study by all techniques of geological and geochemical research, but much controversy still exists regarding the origin of many of the more complex deposits.
The most readily understood ore deposits are those of sedimentary origin. They have been formed at the surface of the Earth by processes that can usually be observed operating at the present time and that can readily be simulated in the laboratory. Salt deposits are one kind whose origin is clearly amenable to such an approach. As long ago as 1849 an Italian scientist initiated laboratory studies on the evaporation of seawater and elucidated the sequence of crystallization of the different salts. Comparison of the results with the mineralogy of salt deposits revealed gross similarities but also important differences; these differences can be explained by a variety of mild metamorphic reactions resulting from burial of these deposits under overlying sediments.
Some sedimentary deposits are not readily explicable by such an approach, however. The most extensive and economically important are the vast Precambrian iron ore deposits, which are a major source for the hundreds of millions of tons of steel produced annually. They occur on all the continents (except perhaps Antarctica) and are uniformly of great age (about 1,900,000,000 years or older). Probably the most extensive and best exposed of these are in the Hamersley Range of Western Australia, where individual beds of iron ore are continuous over hundreds of square miles in a horizontally bedded sequence of iron ore and quartzite thousands of feet thick. The conditions that gave rise to these deposits were apparently unique to this early period in Earth history, because similar deposits are not known in younger geological formations. It has been argued that the explanation lies in an oxygen-free reducing atmosphere in early geological times, under which iron could readily be transported in solution as ferrous compounds to the ocean or large lakes, where deposition eventually took place, perhaps through the agency of primitive organisms. As soon as free oxygen appeared in the atmosphere, 1,000,000,000 to 2,000,000,000 years ago, the geochemical cycle for iron was profoundly modified, and this type of transportation and deposition ended forever.
Processes other than fractional crystallization from igneous melts also give rise to magmatic ore deposits. Economic deposits of the oxide mineral chromite ([Fe,Mg] [Cr,Al]2O4), for example, occur almost entirely as bands or lenses in magnesium-rich igneous rocks. Chromite evidently crystallizes early from a magma, and, being of higher density than the liquid, it sinks to the bottom of the magma chamber and becomes concentrated as almost pure bodies of this mineral. Some accessory minerals of igneous rocks are important sources of metallic elements, but the rocks cannot be mined directly because the grade is too low. If these minerals are chemically and mechanically resistant, weathering and transportation may eventually concentrate them into workable deposits. A large proportion of the world’s zirconium, hafnium, rare earths, and thorium, and some iron and titanium, come from such deposits in river and beach sands.
A large number of important ore deposits occur in metamorphic rocks. The ultimate origin of these deposits is frequently obscured by the complex processes they have undergone. If it can be established that the enclosing metamorphic rocks were of sedimentary origin, the question then arises whether the ore material was deposited along with the sediments or was introduced by circulating solutions during the metamorphism or possibly at some later time. The answer is seldom clear-cut, and such deposits continue to excite lively controversy among geochemists and economic geologists.
Mineral fuels
The mineral fuels—coal, petroleum, and natural gas—may be described as a special type of economic deposit. Geochemically they represent the concentration of carbon and hydrogen by processes that were initially biological in nature. Coal is essentially the product of accumulation of land plants in large amounts, and petroleum and natural gas are the products of marine organisms (although the origin of some petroleum and natural gas under nonmarine conditions cannot be entirely excluded). The origin of petroleum and natural gas presents a more difficult problem than coal because they are fluids and thus are free to migrate from their place of origin.
The formation of coal is a relatively straightforward geochemical process that can readily be traced through its successive stages. The first requirement is a geological one—the rapid accumulation of plant material under conditions that inhibit its decomposition, followed by its burial under inorganic sediments such as shales and sandstones. The great coal-forming period in the Northern Hemisphere followed the Devonian Period (345,000,000 to 395,000,000 years ago), when abundant land plants first appeared, and has been named the Carboniferous Period (280,000,000 to 345,000,000 years ago). During this period, large areas in North America and Europe were evidently low-lying swamps that supported a lush vegetation. This vegetation died, accumulated in successive layers, and was partly decomposed by bacteria and other organisms to form peat. Burial of peat deposits under inorganic sediments brought an end to the period of bacterial decomposition, and the further changes to coal were essentially a mild metamorphism caused by an increase in temperature and pressure.
Chemically, this mild metamorphism was in large part the expulsion of carbon dioxide and water from the coal-forming substance. The main trend in the change from peat through lignite to bituminous coal and anthracite is the decrease in oxygen content and the increase in carbon. If carried to its ultimate conclusion the product would be pure carbon in the form of graphite. This occurs comparatively rarely, but evidence for it is the presence of small amounts of graphite in many metamorphic rocks.
Coal also contains inorganic material that appears as ash when it is burned, and some coal ashes show a remarkable concentration of unusual elements. This was demonstrated by Goldschmidt in 1933, when he found appreciable amounts of germanium in some coal ashes. The Hartley Seam of the Durham Coalfield in England contains so much germanium that the ash has a brilliant yellow colour because of the presence of the oxide (GeO2).
The source of these minor and trace elements and their mode of incorporation in the coal are still not fully understood. There are three possibilities: (1) these elements were taken up by the plants during growth; (2) they were carried into the coal swamp as a component of the inorganic sediments; or (3) they were absorbed during or after the coal-forming processes from circulating solutions. The first possibility is not favoured, because growing plants seldom incorporate appreciable amounts of nonorganic elements. The second possibility also is unlikely, because there is no correlation, or rather an inverse correlation, between ash content and trace element concentrations. This leaves the third possibility as the most likely one. The presence of a large amount of carbonaceous matter means that the coal-forming environment is a highly reducing one, which will favour the precipitation of some elements; the presence of hydrogen sulfide and sulfide ions will cause the precipitation of chalcophile elements (with affinity for sulfur); and complex organic compounds are noted for their absorptive, or chelating, capacity for metallic ions. Thus several individual reactions are potentially available for the fixation of foreign elements in the coal substance.
As pointed out above, the origin of petroleum is not as readily elucidated as the origin of coal, because petroleum can migrate from the region in which it was formed. Indeed, the very occurrence of a commercial oil field probably implies the concentration of the petroleum from a large volume of source rocks into a relatively much smaller reservoir.
The fact that petroleum is almost always found in marine sedimentary rocks has long been a basic argument in favour of a marine origin for this material. It is certainly true that some oil has been found in igneous and metamorphic rocks, but migration from a sedimentary source bed is a reasonable explanation for these occurrences. Proof of a marine origin has been forthcoming in recent years by the sensitive analyses of recent marine sediments, which show that they contain small amounts of petroleum hydrocarbons, evidently generated either directly by marine organisms or by their subsequent decomposition.
Natural petroleum is a complex mixture of hundreds of different hydrocarbons, but its bulk composition is remarkably constant, about 85 percent carbon and 15 percent hydrogen. It may include small amounts of organic compounds containing oxygen, sulfur, and nitrogen. Its content of other elements is exceedingly small. Petroleum ash, unlike coal ash, is not noted for its trace element content. Some petroleum ash contains appreciable amounts of vanadium, however, and has been utilized as a source of this element. A class of nitrogen-bearing organic compounds known as porphyrins include a metal atom in their molecular structure; usually this atom is iron, but other elements in this region of the periodic table, especially vanadium, nickel, and copper, may play a similar role. The vanadium content of some petroleum ash probably originates as a vanadium porphyrin in some of the organisms involved in petroleum formation.
Petroleum is always accompanied by natural gas, but many natural gas fields have no petroleum associated with them. This can probably be ascribed to greater possibility for migration for a gas as compared to a liquid. It is also possible that some natural gas is generated from coal deposits. Natural gas consists largely of methane, but small amounts of more complex hydrocarbons may be present, and it may also contain unwanted components such as nitrogen, carbon dioxide, and hydrogen sulfide. Natural gas containing hydrogen sulfide is known as “sour gas” and for long was an undesirable material because of the noxious nature of this compound; recently, however, it has been found profitable to extract the hydrogen sulfide by converting it to sulfur and then utilize the hydrocarbons.
Natural gas is the sole source of one element, helium, the industrial demand for which has steadily increased in recent years. Comparatively few occurrences of natural gas contain sufficient helium for the extraction to be commercially profitable. Currently, the Western world’s need for helium is largely met by its extraction from wells in western Texas. The explanation for this local concentration of helium in some natural gas fields is still a matter for discussion; the only reasonable source is from the disintegration of radioactive elements in the crust, but the mechanism of concentration remains something of an enigma.
Soils
Soil is a thin veneer that forms a discontinuous cover on the land areas of the Earth. Its volume and its mass are small in comparison to the major geospheres, but it is of vast importance to man. Superficially it might be considered merely as comminuted (pulverized) and decomposed bedrock; however, this viewpoint takes into account only its inorganic components and completely neglects the complex of organic compounds, living organisms, water, and included gases that gives the soil its characteristic properties and its value as the abode of life. Comminuted bedrock alone is not soil in any real sense; the Antarctic continent, where not ice-covered, has a surface of comminuted bedrock, as does the Moon, but neither material can properly be termed soil.
Soils result from the weathering of rocks, and hence their composition might be expected to reflect the composition of the rocks from which they were formed. This is true only in a very broad sense, however. Environmental factors play an important part in soil formation. The same parent rock may give rise to very different soils under different conditions. Climate, topography, vegetation, biological activity, and time are all important factors in determining the nature of a soil. Climate is probably the most important of these, as can be demonstrated by contrasting the soils developed on the same rock type under tropical and temperate conditions. In general, the soil in the humid tropics will be different in texture and composition and much less fertile, as a result of the intense leaching brought about by high rainfall, high temperatures, and the almost complete removal of organic matter by microorganisms.
The complex of inorganic compounds, organic compounds, water, and air that makes up the soil is in a continual state of change. Water tends to dissolve and remove the relatively soluble elements such as calcium, magnesium, sodium, and potassium, and the comparatively insoluble elements—aluminum, iron, and silicon—are thereby relatively enriched in the soil. The enrichment of iron is frequently manifested by a red-brown or yellow-brown colour caused by an accumulation of iron oxides. The most reactive part of the soil is the complex of clay minerals and organic matter, which is largely responsible for its agronomic characteristics. True soil does not exist without the presence of colloidal and organic matter. The relative absence of soils in desert areas reflects the fact that chemical and physical weathering of rocks alone does not necessarily result in soil formation. Most soil processes are directly or indirectly biological in nature. Organisms and organic compounds produced by their vital activities or their decomposition are effective agents for dissolving and extracting many elements from the inorganic constituents of the soil, thereby making them available for plant growth.
Although soils do differ in composition, the range of variation in the major elements is rather small. Minor and trace elements may show considerably greater variability. The importance of certain trace elements in the soil for the healthy growth of plants, and through the plants, of the animals that graze on them, has become increasingly apparent in recent years. Most soils contain these trace elements in sufficient amounts, but when deficiencies are present, puzzling diseases appear, which in the past have rendered large areas of otherwise suitable land unavailable for farming. On a large area in the North Island of New Zealand, for example, although it grew satisfactory pasture, sheep and cattle failed to thrive and eventually died if not removed. As a result, much of this area was given over to afforestation. It was eventually discovered that cobalt, in the amount of a few parts per million, would completely eliminate the disease when applied in fertilizer or administered directly to the animals. The ultimate explanation is the need of animals (but not plants) for vitamin B12, which contains an atom of cobalt in its structure.
Occasionally, an excess of a specific element may have a deleterious effect on plant growth. Most obvious, of course, are the alkaline or saline soils of desert and coastal areas on which only an impoverished vegetation exists. Magnesium-rich soils are notably infertile; such soils develop on areas of ultrabasic igneous rocks consisting largely of olivine, (Mg,Fe)2SiO4, and the boundaries of these areas can frequently be readily mapped from aerial photographs by the marked change in vegetation. Sometimes plants take up available trace elements in amounts deleterious to animals grazing on them. A well-known example is Astragalus racemosus (locoweed), which in some areas of the western U.S. contains sufficient selenium to be poisonous to grazing animals. The possible correlation between soil geochemistry and the geographical distribution of disease is thus a field of extreme significance which as yet has been insufficiently studied. The problem is a complex one, in large part because of the difficulty in isolating the numerous factors involved.
The hydrosphere
The hydrosphere is the discontinuous shell of water—fresh, salt, and solid—on the surface of the Earth. As such it comprises the oceans and the connecting seas and inlets, the lakes, rivers, and streams, the groundwater that feeds them, and the snow and ice cover of high altitudes and high latitudes. The mass of the ocean waters far outweighs the other parts of the hydrosphere. Goldschmidt estimated that there are 273 litres of water in all its forms for every square centimetre of the Earth’s surface made up as follows:

Seawater thus makes up over 98 percent of the total mass of the hydrosphere, and its composition (see Table) essentially can be taken as giving the average composition of the hydrosphere.
Composition of seawater
Research during the past century has demonstrated that the composition of seawater is essentially uniform and that the relative proportions of the various ions are practically constant. In the open ocean the salinity (approximately the total weight of dissolved solids per kilogram) averages about 35 parts per thousand, but may rise to 40 parts per thousand in regions such as the Red Sea and the Persian Gulf, where rainfall and inflow are low and evaporation high. Sodium chloride is the dominant compound of the salts in solution and comprises about three-quarters of the whole; the remainder consists largely of chlorides and sulfates of magnesium, calcium, and potassium.
Though many data on minor and trace elements in seawater (Table) are available, the interpretation of these data is subject to some uncertainties. The concentrations of these elements are probably more variable than for the major elements and may depend to some degree on the sampling location. This is particularly true for the elements that are utilized by marine organisms. Phosphorus is a good example; it is markedly depleted in surface and near-surface waters by biological activity, but it enriches the deeper parts of the ocean through the dissolution of dead organisms. Silica is brought into the ocean in large amounts in solution in river water, but most of it is soon removed to become the skeletal material of diatoms, radiolaria, and sponges. Seawater also contains dissolved gases in variable amounts. Seawater of normal salinity at 0° C (32° F) in equilibrium with the atmosphere will contain about eight millilitres per litre of dissolved oxygen and 14 millilitres per litre of dissolved nitrogen. Dissolved nitrogen is essentially an inert constituent, but dissolved oxygen plays a fundamental role in the growth and decay of organisms and so varies greatly in concentration from place to place. In stagnant regions that are rich in decaying organic matter the water may be completely depleted in free oxygen, and a considerable concentration of hydrogen sulfide may be present. Much of the Black Sea below a depth of a few hundred metres is in this condition.
Another dissolved gas of prime importance for biological activity is carbon dioxide. The conditions controlling its concentration are quite complex, however, because in solution it can be present as free carbon dioxide, as undissociated carbonic acid, as carbonate ions, and as bicarbonate ions. The concentration of these ions will also be affected by biological activity and by the precipitation or dissolution of calcium carbonate.
Circulation of water through the hydrosphere
The circulation of water through the hydrosphere is controlled in large part by the reservoir effect of the oceans. Evaporation from the ocean surface is precipitated as rainfall. Of that falling on the land, some is directly re-evaporated, some is absorbed into the reservoir of groundwater, and some flows off directly into rivers and streams. The total annual rainfall on the Earth is estimated to be 123 × 1018 grams, and the total annual runoff to the oceans 32 × 1018 grams.
Even the purest rainfall contains some material in solution, not only dissolved gases but also nonvolatile material. Rainfall near seacoasts always contains some sodium chloride and small amounts of other marine salts, the concentration of which falls off generally with distance from the ocean. Rainfall in industrial regions may of course contain a variety of pollutants; in many areas it is essentially a dilute sulfuric acid solution. Such material may also be carried far beyond the place of origin; acid rainfall in the Scandinavian countries probably originates in part from England and Germany.
The runoff from the land contains additional material in solution, picked up during its circulation through the crustal rocks. River water averages about 120 parts per million dissolved solids, but the range is great, from about 10 parts per million up to several thousand parts per million. Commonly, the range is from 50 to 200 parts per million; contents greater than 200 parts per million are usually the result of human activities or of drainage from soils containing soluble salts, as in desert regions.
With an average content of 120 parts per million dissolved matter, the rivers of the world deliver 3.9 × 109 tons of material in solution to the sea each year. The average concentration of the important constituents (in parts per million) is: bicarbonate, 58.4; sulfate, 11.2; chloride, 7.8; nitrate, 1.0; calcium, 15.0; magnesium, 4.1; sodium, 6.3; potassium, 2.3; iron, 0.67; and silica, 13.1. Although these ten constituents account for most of the dissolved material, many other elements have been detected in river and lake waters.
Geochemical balance of seawater over time
The 3.9 × 109 tons carried annually in solution to the oceans are but a small fraction of the total amount of material in solution in the oceans. Nevertheless, when integrated over the whole of geological time, more than 4 × 109 years, it greatly exceeds the present material in solution. Some of the material, especially sodium chloride, is of course cyclical, being circulated from the oceans to the land as aerosols and incorporated in marine sedimentary rocks and ultimately in large part being returned to the oceans in runoff.
Goldschmidt made an interesting calculation on the geochemical balance in seawater. From the amount and composition of sedimentary rocks he estimated that erosion during geological time had amounted to about 160 kilograms of igneous rock per square centimetre of the Earth’s surface. Combining this figure with the amount of seawater per square centimetre, 273 kilograms, he derived a figure of 600 grams of igneous rock eroded per kilogram of seawater. Assuming this 600 grams had gone fully into solution (obviously a gross simplification but a limiting one), he drew up a balance sheet between the amounts of different elements potentially supplied to the oceans and the amounts actually present. Some of these figures are presented in the Table. Despite the imperfections of the method, the results are certainly significant in a qualitative sense. Some elements—chlorine, bromine, boron, and sulfur—are present in seawater in amounts far in excess of those that can have been derived by erosion. The source of these “superabundant” elements has probably been volcanism and related magmatic activity. Halides, sulfates, and borates are deposited by volcanic gases and carried in solution in hot springs. The relative depletion of fluorine with respect to chlorine in seawater can be ascribed to the precipitation of highly insoluble fluorine-bearing compounds, mainly apatite (calcium fluophosphate). Sodium clearly remains in solution to a much greater extent than potassium; the latter element reacts with sedimentary materials to form insoluble potassium-bearing silicates such as illite and glauconite, which have no sodium-bearing analogs. Calcium is removed from solution much more effectively than strontium, evidently because it is utilized by organisms. Goldschmidt pointed out that many highly poisonous elements, such as arsenic and selenium, have been potentially supplied in dangerous amounts. Their concentration remains very low, however, presumably because of efficient processes of removal as insoluble compounds. Adsorption on colloidal particles of clay and iron oxides is a likely process.
| Geochemical balance of some elements in seawater | |||
| element | potential amount supplied to oceans (g/ton) | amount present in seawater (g/ton) | percentage in solution |
| lithium | 39 | 0.17 | 0.4 |
| boron | 2 | 4.5 | 250 |
| fluorine | 540 | 1.3 | 0.2 |
| sodium | 16,980 | 10,800 | 64 |
| magnesium | 12,540 | 1,290 | 10 |
| phosphorus | 708 | 0.09 | 0.01 |
| sulfur | 312 | 904 | 290 |
| chlorine | 188 | 19,400 | 10,300 |
| potassium | 15,540 | 392 | 2.5 |
| calcium | 21,780 | 411 | 1.9 |
| arsenic | 3 | 0.003 | 0.1 |
| bromine | 0.97 | 67 | 6,900 |
| rubidium | 186 | 0.12 | 0.06 |
| strontium | 180 | 8.1 | 4.6 |
| iodine | 0.18 | 0.06 | 33 |
| cesium | 4 | 0.0003 | 0.008 |
| barium | 150 | 0.02 | 0.01 |
The geological and geochemical evidence indicates that the ocean waters are, and have been for a long time, in a steady state of essentially unchanging composition. The addition of material by runoff from the land is adjusted by reactions within the ocean waters or between the ocean waters and sedimentary materials whereby the concentrations of the individual elements remain essentially constant. How far back in geological time this steady state has persisted remains something of an open question. The existence of most forms of marine life from the Cambrian to the present indicates a uniformity of marine conditions over the past 600,000,000 years; how far back into the Precambrian this uniformity extended is more difficult to elucidate. The earlier discussion of Precambrian iron formations suggested the possibility of very different atmospheric composition some 2,000,000,000 years ago, and the considerable interdependence of atmospheric and oceanic composition indicates that this might have resulted in marked geochemical differences in the ocean waters.
The atmosphere
The atmosphere is the most homogeneous and thus the most easily studied of the geospheres. Its mass is readily determined from the product of the average height of the mercury barometer in centimetres, the density of mercury (13.6 grams per cubic centimetre), and the area of the Earth (5.1 × 1018 square centimetres). Recent calculations give 51.17 × 1020 grams for its total mass.
Composition
The composition is also relatively simple, although a considerable number of gases may be present in small amounts (Table). Almost 99 percent consists of oxygen and nitrogen, with argon making up most of the remainder. Carbon dioxide, essential for plant life, is present in an extremely small amount. Some gases not listed in the Table may be present as local or even regional pollutants—city dwellers are becoming increasingly aware of oxides of sulfur as atmospheric pollutants, and the scientific study of smog is largely concerned with reactions taking place between hydrocarbons, oxides of nitrogen, oxygen and ozone.
| Average composition of the atmosphere | |||
| *ppm = parts per million. **Variable, increases with height. | |||
| gas | composition by volume (ppm)* | composition by weight (ppm)* | total mass (1020 g) |
| nitrogen | 780,900 | 755,100 | 38.648 |
| oxygen | 209,500 | 231,500 | 11.841 |
| argon | 9,300 | 12,800 | 0.655 |
| carbon dioxide | 386 | 591 | 0.0299 |
| neon | 18 | 12.5 | 0.000636 |
| helium | 5.2 | 0.72 | 0.000037 |
| methane | 1.5 | 0.94 | 0.000043 |
| krypton | 1.0 | 2.9 | 0.000146 |
| nitrous oxide | 0.5 | 0.8 | 0.000040 |
| hydrogen | 0.5 | 0.035 | 0.000002 |
| ozone** | 0.4 | 0.7 | 0.000035 |
| xenon | 0.08 | 0.36 | 0.000018 |
The atmosphere gradually thins out into the vacuum of outer space, and its upper limit can conveniently be placed at about 600 kilometres. An important zone in the stratosphere is known as the ozonosphere, a diffuse layer characterized by an increase in the concentration of ozone, O3. This zone is highly important for life on Earth because it absorbs most of the ultraviolet radiation from the Sun; if this penetrated to the Earth’s surface it would act as a potent sterilizer, fatal for most forms of life. It also helps to maintain a more uniform surface temperature by reducing the loss of heat by radiation to space—the so-called greenhouse effect.
Geochemical history
The geochemical history of the atmosphere has been a complex one. Scientists agree that the present atmosphere is quite different from the original one. It is certainly quite different from those of the other planets. It is reasonable to conclude that this reflects, in part at least, the Earth as the abode of life. The Earth’s atmosphere differs from those of its neighbours in the solar system probably in large part through the action of photosynthesis, a complex biological process which was probably preceded by a lengthy period of organic evolution.
The nature of the Earth’s primitive atmosphere is still a subject of some speculation. Some scientists, reasoning by analogy with the larger planets such as Jupiter, have argued for an original atmosphere consisting largely of methane and ammonia. Others have considered that present-day volcanic gases may indicate the nature of the primitive atmosphere, in which case it contained carbon dioxide, possibly carbon monoxide, nitrogen, and water vapour. In either case, free oxygen was absent. If the evolution of the atmosphere is traced backward in time through the geological record, then extensive terrestrial photosynthesis is indicated by an abundance of land plants in Devonian times, about 400,000,000 years ago. Marine photosynthesis, however, is much older, since practically all the major groups of marine organisms were established by the beginning of the Cambrian period, some 540,000,000 years ago. As discussed earlier, the extensive Precambrian iron formations suggest an oxygen-free atmosphere which was terminated about 2,000,000,000 years ago. This evidence has been translated into estimates of oxygen content of the atmosphere of about 1 percent of the present level 2,000,000,000 years ago, about 10 percent of the present level at the beginning of the Cambrian Period, and essentially the present content by Devonian times.
Although it is not yet possible to know the quantitative composition of the primitive atmosphere, the geochemical processes that have operated to modify its composition during geological time can be evaluated. These processes can be summarized as a series of gains and losses. Additions to the atmosphere comprise: (1) gases released by igneous activity; (2) oxygen and hydrogen produced by the photochemical dissociation of water vapour; (3) oxygen produced by photosynthesis; (4) helium produced by the radioactive breakdown of uranium and thorium; and (5) argon produced by the radioactive breakdown of potassium. Atmospheric losses include: (1) oxygen removal by oxidation of ferrous to ferric iron, sulfur compounds to sulfates, hydrogen to water, and similar reactions; (2) carbon dioxide removed by the formation of coal, petroleum, and the death and burial of organisms; (3) carbon dioxide removed by the formation of calcium and magnesium carbonates; (4) nitrogen removed by the formation of oxides of nitrogen in the air and by the action of nitrifying bacteria in the soil; and (5) hydrogen and helium by escape from the Earth’s gravitational field.
Photosynthesis has certainly been the most significant process in controlling atmospheric composition during much of geological time. Through this process carbon dioxide and water are converted to carbohydrate, with the accompanying release of oxygen. Much of this carbohydrate is consumed by animals and reconverted to carbon dioxide and water by respiration, and oxidative decay leads to the same result. Some, however, is incorporated into sediments; part may go to form exploitable deposits of coal and petroleum, but most of it remains as disseminated carbonaceous material; the average carbon content of sedimentary rocks is about 0.4 percent.
Quantitatively, more significant amounts of carbon dioxide have been removed from the atmosphere in the form of limestone and dolomite. Most of this removal has been effected by marine organisms, especially algae and corals, but direct inorganic precipitation may occur, especially in warm tropical waters. Judging from the vast deposits of limestone and dolomite throughout the sedimentary record, this process has operated with a remarkable degree of uniformity throughout geological time. Extensive limestone formations are possibly less common in older Precambrian rocks, indicating a slower beginning of carbonate precipitation. It is truly remarkable that so much carbonate rock has been deposited during geological time, with the carbonate being ultimately derived from an atmosphere which may never have contained a much higher concentration of carbon dioxide than is present today. It has been pointed out that reactions like the decomposition of calcium silicate—CaSiO3 + CO2 = CaCO3 + SiO2, in which CaSiO3 is calcium silicate, CO2 is carbon dioxide, CaCO3 is calcium carbonate, and SiO2 is silicon dioxide, which tends to go toward the right at ordinary temperatures—will act as buffering mechanisms to keep the carbon dioxide concentration of the atmosphere at a continuously low level.
If the carbon dioxide concentration has remained essentially constant, and yet this compound has been continuously extracted to form carbonates and organic compounds, then clearly a balancing source of “new” carbon dioxide is required. This evidently has been provided by volcanism and other igneous activity. The Earth is steadily being degassed, in the sense that gaseous compounds contained in the mantle are escaping to the surface. The presence of carbon dioxide in the mantle has been demonstrated by the presence of microscopic inclusions of liquid carbon dioxide in the minerals of the peridotite xenoliths (rocks contained within other rocks) brought up in some volcanoes. Along with carbon dioxide, much water and small amounts of other volatiles are being added continuously from sources in the mantle. Ultimately, the hydrosphere, as well as the atmosphere, is the product of the degassing of the Earth’s interior.
Of the remaining atmospheric gases, argon presents some intriguing features. Argon is by far the most abundant of the inert gases on Earth, whereas in the universe as a whole it is much less abundant than either helium or neon. In addition, its isotopic composition is quite distinct, consisting almost entirely of argon-40, whereas in the Cosmos argon-36 is the most abundant isotope. The reason for these anomalies is that atmospheric argon is almost entirely radiogenic, the product of the decay of the potassium-40 isotope of potassium.
Similarly, the helium in the atmosphere is probably entirely the product of the radioactive decay of uranium and thorium. Actually, the atmosphere contains only about 10 percent of the total amount of helium generated from these sources during geological time. Some of this helium remains occluded in the rocks where it was formed, some has escaped from the upper atmosphere. Helium (and hydrogen), consisting of light atoms, can escape from the gravitational field of the Earth, whereas heavier gases cannot. A minor source of atmospheric oxygen throughout geological time is probably the photochemical decomposition of water vapour in the upper atmosphere, with the subsequent loss of the hydrogen to outer space.
Some oxygen has been removed from the atmosphere by oxidative reactions, of which the most significant has been the conversion of ferrous to ferric iron. In igneous rocks the average ferrous-to-ferric iron ratio (FeO/Fe2O3) is greater than unity, whereas in sedimentary rocks the proportion is reversed, ferric iron being dominant over ferrous iron. Other oxidative reactions are the conversion of manganous compounds to manganese dioxide and of hydrogen sulfide to free sulfur and sulfate. Nitrogen is almost inert geochemically, but a little is fixed as oxides of nitrogen by lightning, and somewhat more by the action of nitrifying bacteria in the soil. Most of this nitrogen is ultimately returned to the atmosphere by the decay of the organisms. Oxides of nitrogen formed in the atmosphere are removed in rain as nitrite and nitrate. Nitrogen does not accumulate in the soil, however, except perhaps under extremely arid conditions, as in the deserts of northern Chile, the locale of the unique nitrate deposits.