Chemical kinetics, the branch of physical chemistry that is concerned with understanding the rates of chemical reactions. It is to be contrasted with thermodynamics, which deals with the direction in which a process occurs but in itself tells nothing about its rate. Thermodynamics is time’s arrow, while chemical kinetics is time’s clock. Chemical kinetics relates to many aspects of cosmology, geology, biology, engineering, and even psychology and thus has far-reaching implications. The principles of chemical kinetics apply to purely physical processes as well as to chemical reactions.
One reason for the importance of kinetics is that it provides evidence for the mechanisms of chemical processes. Besides being of intrinsic scientific interest, knowledge of reaction mechanisms is of practical use in deciding what is the most effective way of causing a reaction to occur. Many commercial processes can take place by alternative reaction paths, and knowledge of the mechanisms makes it possible to choose reaction conditions that favour one path over others.
A chemical reaction is, by definition, one in which chemical substances are transformed into other substances, which means that chemical bonds are broken and formed so that there are changes in the relative positions of atoms in molecules. At the same time, there are shifts in the arrangements of the electrons that form the chemical bonds. A description of a reaction mechanism must therefore deal with the movements and speeds of atoms and electrons. The detailed mechanism by which a chemical process occurs is referred to as the reaction path, or pathway.
The vast amount of work done in chemical kinetics has led to the conclusion that some chemical reactions go in a single step; these are known as elementary reactions. Other reactions go in more than one step and are said to be stepwise, composite, or complex. Measurements of the rates of chemical reactions over a range of conditions can show whether a reaction proceeds by one or more steps. If a reaction is stepwise, kinetic measurements provide evidence for the mechanism of the individual elementary steps. Information about reaction mechanisms is also provided by certain nonkinetic studies, but little can be known about a mechanism until its kinetics has been investigated. Even then, some doubt must always remain about a reaction mechanism. An investigation, kinetic or otherwise, can disprove a mechanism but can never establish it with absolute certainty.
Reaction Rate
The rate of a reaction is defined in terms of the rates with which the products are formed and the reactants (the reacting substances) are consumed. For chemical systems it is usual to deal with the concentrations of substances, which is defined as the amount of substance per unit volume. The rate can then be defined as the concentration of a substance that is consumed or produced in unit time. Sometimes it is more convenient to express rates as numbers of molecules formed or consumed in unit time.
The half-life
A useful rate measure is the half-life of a reactant, which is defined as the time that it takes for half of the initial amount to undergo reaction. For a special type of kinetic behaviour (first-order kinetics; see below Some kinetic principles), the half-life is independent of the initial amount. A common and straightforward example of a half-life independent of the initial amount is radioactive substances. Uranium-238, for example, decays with a half-life of 4.5 billion years; of an initial amount of uranium, half of that amount will have decayed in that period of time. The same behaviour is found in many chemical reactions.
Even when the half-life of a reaction varies with the initial conditions, it is often convenient to quote a half-life, bearing in mind that it applies only to the particular initial conditions. Consider, for example, the reaction in which hydrogen and oxygen gases combine to form water; the chemical equation is2H2 + O2 → 2H2O.If the gases are mixed together at atmospheric pressure and room temperature, nothing observable will happen over long periods of time. However, reaction does occur, with a half-life that is estimated to be more than 12 billion years, which is roughly the age of the universe. If a spark is passed through the system, the reaction occurs with explosive violence, with a half-life of less than one-millionth of a second. This is a striking example of the great range of rates with which chemical kinetics is concerned. There are many possible processes that proceed too slowly to be studied experimentally, but sometimes they can be accelerated, often by the addition of a substance known as a catalyst. Some reactions are even faster than the hydrogen-oxygen explosion—for example, the combination of atoms or molecular fragments (called free radicals) where all that occurs is the formation of a chemical bond. Some modern kinetic investigations are concerned with even faster processes, such as the breakdown of highly energetic and therefore transient molecules, where times of the order of femtoseconds (fs; 1 fs = 10–15 second) are involved.
Measuring slow reactions
The best way to study exceedingly slow reactions is to change the conditions so that the reactions occur in a reasonable time. Increasing the temperature, which can have a strong effect on the reaction rate, is one possibility. If the temperature of a hydrogen-oxygen mixture is raised to about 500 °C (900 °F), reaction then occurs rapidly, and its kinetics has been studied under those conditions. When a reaction occurs to a measurable extent over a period of minutes, hours, or days, rate measurements are straightforward. Amounts of reactants or products are measured at various times, and the rates are readily calculated from the results. Many automated systems have now been devised for measuring rates in this way.

Chemical kinetics
NOTABLE HONOREES
- Sir Cyril Norman Hinshelwood
RELATED TOPICS
- Reaction rate
- Physical chemistry
Measuring fast reactions
Some processes are so fast that special techniques have to be used to study them. There are two difficulties with fast reactions. One is that the time that it takes to mix reactants or to change the temperature of the system may be significant in comparison with the half-life, so that the initial time cannot be measured accurately. The other difficulty is that the time it takes to measure the amounts of substances may be comparable with the half-life of the reaction. The methods used to overcome these difficulties fall into two classes: flow methods and pulse and probe methods.
In flow methods, two gases or solutions are introduced rapidly into a mixing vessel, and the resulting mixture then flows rapidly along a tube. Concentrations of reactants or products may then be measured—for example, by spectroscopic methods—at various positions along the tube, which correspond to various reaction times. A modification of this method is the stopped-flow technique, in which the reactants are forced rapidly into a reaction chamber; the flow is then suddenly stopped, and the amounts are measured by physical methods after various short times. These flow methods are limited by the time it takes to mix gases or solutions and are not suitable if the half-life is less than about a hundredth of a second.
These mixing difficulties were overcome by pulse and probe methods. The principle of these is that a short pulse, usually of radiation, is given to a chemical system and is then followed by a probe, usually involving radiation that provides spectroscopic evidence of what occurred after the initial pulse. The first of these methods, developed in 1949 by British chemists R.G.W. Norrish and George Porter, was the flash-photolysis method, for which Norrish and Porter won the Nobel Prize for Chemistry in 1967. In this technique a flash of light of high intensity but short duration brings about the formation of atomic and molecular species, the reactions of which can be studied kinetically by spectroscopy. In the earliest experiments the duration of the flash was about a millisecond (ms; 1 ms = 10–3 second), but in the next four decades the duration was reduced by more than 11 powers of 10, to just a few femtoseconds. A nanosecond (ns; 1 ns = 10–9 second) flash is adequate for studying almost any purely chemical reaction where there is a change in chemical identity. Any chemical reaction, however, involves processes of a purely physical nature, such as energy redistribution and the breakdown of transient species, which occur in the femtosecond range.
Many such processes have now been studied with flashes of only a few femtoseconds’ duration. The time that it takes for the length of a chemical bond to change by 10–10 metre can be as little as about 100 fs, so that a flash of a few femtoseconds’ duration, closely followed by another one of the same duration, will provide information about such tiny changes in bond lengths. The technique for causing one flash to occur a few nanoseconds after another is to route the light by a slightly longer path. A path of 1 additional micrometre (μm; 1 μm = 10–6 metre) causes a delay of 1 fs, and such a short path difference is now technically feasible. Egyptian-born chemist Ahmed Zewail won the Nobel Prize for Chemistry in 1999 for his work in this field.
Another pulse method is the relaxation method, developed in the 1950s by German physicist Manfred Eigen (who shared the Nobel Prize for Chemistry in 1967 with Norrish and Porter). In this method the investigation begins with a reaction system in equilibrium; the reaction to be studied has finished, and no further changes take place. The external conditions are then altered very rapidly; the system is then no longer at equilibrium, and it relaxes to a new equilibrium. The speed of relaxation is measured by a physical method such as spectroscopy, and analysis of the results leads to the reaction rate.
The most common way of changing the external conditions is to change the temperature, and the method is called the temperature-jump, or T-jump, method. Techniques have been developed for raising the temperature of a tiny reaction vessel by a few degrees in less than 100 ns. The method is therefore not suitable for the fastest processes, which can be studied by flash photolysis, but many purely chemical processes are suitable for the T-jump technique, which has provided valuable kinetic information. (See also relaxation phenomenon.)
Other experimental techniques are used for the study of rapid processes. Ultrasonic methods have been used for processes occurring with half-lives in the microsecond (μs; 1 μs = 10−6 second) and nanosecond ranges. Nuclear magnetic resonance has also been used for certain types of reactions.

SIMILAR TOPICS
· Electrochemistry
· Stereochemistry
Some Kinetic Principles
The kinetic behaviour of an ordinary chemical reaction is conventionally studied in the first instance by determining how the reaction rate is influenced by certain external factors such as the concentrations of the reacting substances, the temperature, and sometimes the pressure. For a reaction in which two substances A and B react with each other, it is sometimes found that the reaction rate is proportional to the concentration of A, represented by [A], and to the concentration of B, or [B]. In that case the reaction is said to be a second-order reaction; it is first order in [A] and first order in [B]. In such a case the reaction rate v can be expressed asv = k[A][B],where k is a constant, known as the rate constant for the reaction.
READ MORE ON THIS TOPIC
ion-exchange reaction: Ion-exchange kinetics
Generally, ion exchange is fast. No electron-pair bonds need be broken, and the rate of the process is limited only by the rate at which…
This is just one of many types of kinetics that can be observed. A substance A that changes into another substance may obey a kinetic equation of the form v = k[A], which is a first-order reaction. It is important to recognize that the kinetics of a reaction does not always correspond in a simple way to the balanced chemical equation for the reaction. Thus, if a reaction is of the formA + B ⇌ Y + Z,the reaction is not necessarily second-order in both directions. This is in contrast to the situation with the equilibrium constant for the reaction, which corresponds to the balanced equation. The reason why the kinetic law is different is that the reactions in the forward and reverse directions may occur by stepwise mechanisms that lead to a different and usually more complex kinetic equation.
Sometimes reaction rates depend on reactant concentrations in a more complicated way. This is a clear indication that a reaction happens in several steps (see below Composite reaction mechanisms).
The effect of temperature on reaction rates provides much information about reaction mechanisms. Understanding of this effect owes much to the ideas of the Dutch physical chemist Jacobus Henricus van ’t Hoff and the Swedish chemist Svante August Arrhenius. Their equation for the dependence of a rate constant k on the absolute temperature T isk = A exp (−E/RT),where R is the molar gas constant and A and E are quantities that are different for each reaction. This equation has come to be called the Arrhenius equation, although, as Arrhenius acknowledged when he applied it in 1889, it was first suggested by van ’t Hoff in 1884. According to this relationship, a plot of the logarithm of the rate constant against the reciprocal of the absolute temperature should yield a straight line. From the slope and intercepts of the line, it is possible to calculate the value of the kinetic parameters A and E. The Arrhenius relationship applies satisfactorily to most reactions and indeed to many physical processes; however, various complications may cause it to fail.
If the reaction between two molecules is an elementary one, occurring in a single step, a simple interpretation of the Arrhenius equation can be given. The quantity A is related to the frequency of collisions between the reacting molecules. The quantity E, known as the activation energy for the reaction, results from the fact that there is an energy barrier to reaction. If E was zero, k would be equal to A, which means that the reaction would occur every time a collision occurred between the reactant molecules. This is the case for reactions in which no chemical bond is broken, such as the combination of atoms.
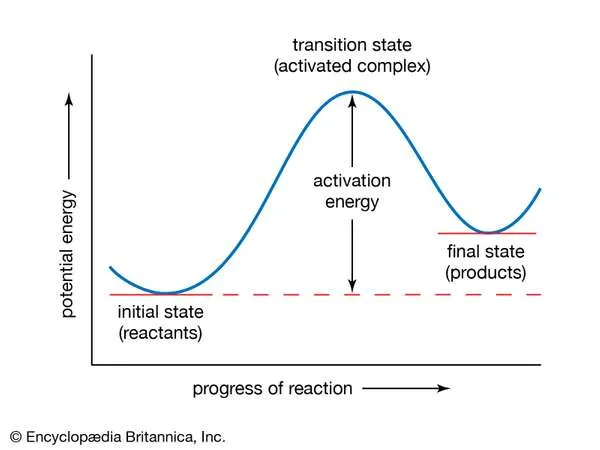
In the Arrhenius equation, the activation energy (E) represents the minimum amount of energy required to transform reactants into products in a chemical reaction. On a potential energy curve, the value of the activation energy is equivalent to the difference in potential energy between particles in an intermediate configuration (known as the activated complex, or transition state) and particles of reactants in their initial state. The activation energy thus can be visualized as a barrier that must be overcome by reactants before products can be formed.Encyclopædia Britannica, Inc.
For reactions in which a chemical bond is broken, on the other hand, the activation energy E is not zero but has a value that is often a tenth or so of the energy required to break the bond. A simple and essentially correct explanation of the activation energy was suggested by Arrhenius, who pointed out that, for many reactions, raising the temperature by 10 °C (18 °F) doubles the reaction rate. This increase cannot be caused by the increase in the frequency of collisions between colliding molecules, since the frequency does not increase sufficiently with a rise in temperature. Arrhenius suggested that when reactants A and B react together, they first form a highly energized intermediate that is denoted as AB‡, which subsequently gives the products of reaction:A + B ⇌ AB‡ → Y + Z.
If the intermediate complex (also called the activated complex) AB‡ is of high energy, it is formed only in small amounts. According to the Boltzmann principle, the fraction of molecules having energy greater than E is exp (−E/RT), which provides an explanation of the appearance of this fraction in the Arrhenius equation. The interpretation of the equation is thus that only those molecules having energy greater than E are able to undergo reaction; other collisions are ineffective, and the reactant molecules merely separate unchanged. (See below Theories of reaction rates.)
Composite Reaction Mechanisms
Various lines of evidence are used to determine if a reaction occurs in more than one step. Suppose that the kinetic equation for the reaction does not correspond to the balanced equation for the reaction. A simple example is the reaction between hydrogen and iodine chloride, with the formation of iodine and hydrogen chloride:H2 + 2ICl → I2 + 2HCl.
To make the equation balance, the reaction must be written as shown, with two iodine chloride molecules reacting with a single hydrogen molecule. If this reaction occurred in a single elementary step, the rate would be proportional to the first power of the hydrogen concentration and the square of the iodine chloride concentration. Instead, however, the rate is found to be proportional to both concentrations to the first power, so that it is a second-order reaction:v = k[H2][ICl].This can be explained if there is initially a slow reaction between one hydrogen molecule and one of iodine chloride:H2 + ICl → HI + HCl (slow)followed by a rapid reaction between the hydrogen iodide formed and an additional molecule of iodine chloride:HI + ICl → HCl + I2 (fast).
If the second reaction is fast, the hydrogen iodide is removed as fast as it is formed. The rate of the second reaction therefore has no effect on the overall rate, which is the rate of the first step. This mechanism therefore explains the kinetic behaviour but does not prove it; other, more complicated schemes could be devised, but, until there is further evidence, it is expedient to accept the simple mechanism. This is an example of a consecutive reaction, which occurs in two steps, with the intermediate playing a role.
Another piece of evidence for a composite mechanism is the detection of reaction intermediates. In such a case, a reaction scheme must be devised that will account for these intermediates. Sometimes an intermediate can be a fairly stable substance. In other cases the intermediates are unstable species such as atoms and free radicals (fragments of molecules) that subsequently undergo rapid reactions. Free radicals can be detected by spectroscopy and other means. When organic molecules are raised to high temperatures, they decompose into smaller molecules, and organic free radicals have often been detected as intermediates. In an explosion, such as that between hydrogen and oxygen, free radicals such as hydroxyl can be detected.
Composite reaction mechanisms are of various kinds. Aside from the simple consecutive schemes, there are some special mechanisms that give rise to oscillatory behaviour: the amount of a product continuously rises and falls over a period of time. The conditions for this behaviour are that there must be at least two species involved in the reaction and there must be feedback, which means that products of the reaction affect the rate. There are also reaction mechanisms that give rise to what is technically known as chaos, or catastrophe. With such reactions it is impossible to predict the outcome. Chaotic conditions also require that there be feedback and that at least three species be involved.
Sometimes a complex reaction mechanism involves a cycle of reactions such that certain intermediates consumed in one step are regenerated in another. For example, the accepted mechanism of the reaction between hydrogen and bromine, which can be written asH2 + Br2 → 2HBr,includes the stepsBr + H 2 → HBr + HH + Br 2 → HBr + Br.
In the first of these steps a bromine atom is consumed, but in the second a bromine atom is regenerated. This pair of reactions can thus occur with the production of two molecules of hydrogen bromide, the product of the reaction, without loss of bromine atoms. This pair of reactions is called a cycle of reactions, and it can occur a number of times, in which case the reaction is referred to as a chain reaction. The two reactions in which bromine is regenerated are known as the chain-propagating steps. The average number of times the pair of steps is repeated is known as the chain length.
One necessary condition for a proposed reaction mechanism to be correct is that it must account for the overall kinetic behaviour of the reaction—in particular, for the dependence of the reaction rate on the reactant concentrations. For any proposed reaction mechanism, it is possible to write down equations for the rate of each step in terms of the reactant concentration and then to solve the equations for the overall rate. A practical difficulty arises, since no exact mathematical solution is possible for all except the simplest of mechanisms. If one has values for the rate constants, solutions can be obtained with a computer, but explicit rate equations provide more insight into the reactions. One therefore looks for approximate solutions of the equations. One of these is provided by the steady-state treatment, which is applicable if (and only if) the intermediates are species that can be present only at low concentrations. If this condition is satisfied by an intermediate, the rate of change of its concentration during the course of reaction is always small and, as a good approximation, can be assumed to be zero, which means that the intermediate exists in a steady state. This approximation may safely be applied to atoms and free radicals present as reaction intermediates. With this approximation it is usually possible to obtain a reliable approximate equation for the overall reaction rate in terms of reactant concentrations. If this agrees with the experimental behaviour, the mechanism is accepted.
One situation to which the steady-state treatment does not apply is when a reaction is an explosion. Explosions occur because the concentration of intermediates does not remain steady during the course of reaction but rises to a high value so that the reaction goes out of control. This occurs if the reaction mechanism involves a special kind of chain called a branching chain. In the hydrogen-oxygen explosion, for example, the following reaction is known to occur:H + O2 → OH + O.In this step a single chain carrier hydrogen atom has produced two chain carriers: a hydroxyl group and an oxygen atom. The number of chain carriers increases rapidly and leads to an explosion.
Theories Of Reaction Rates
Two different theoretical approaches to chemical kinetics have led to an understanding of the details of how elementary chemical reactions occur. Both of these are based on the idea of potential-energy surfaces, which are models showing how the potential energy of a reaction system varies with certain critical interatomic distances. The course of an elementary reaction is represented by the movement of the system over the potential-energy surface. One theoretical approach to the problem involves studying the region of the potential-energy surface that corresponds to the highest point on the energy barrier that separates the reactants from the products. This approach is relatively simple and leads to explicit general expressions for the reaction rate. The second approach involves considering the dynamics of the motion of the system over the potential-energy surface.


{kind=link}